Strains and Growth Conditions
Yeast Strains
Yeast Deletion Strains (purchased from Research Genetics)
BY4741 MATa his3D1 leu2D0 met15D0 ura3D0
| Gene Name(s) | ORF | Function |
| Gcr3, Sto1, Cbc1 | YMR125w | Nuclear cap binding complex subunit |
| Mud13, Cbc2 | YPL178w | Nuclear cap binding complex subunit |
| Nam8, Mre2, Mud15 | YHR086w | U1 snRNP Protein |
| Mud1 | YBR119w | U1 snRNP A protein |
| Mud2 | YKL074c | commitment complex |
| Msl1, Yib1 | YIR009w | U2 snRNP B" protein |
| Cus2 | YNL286w | U2 snRNP |
| Snu17, Ist3 | YIR005w | U2 snRNP protein |
| Snu40, Lin1 | YHR156c | tri-snRNP |
| Snu66 | YOR308c | tri-snRNP |
| Ecm2, Slt11 | YBR065c | Assoc. with U2, second step factors |
| Prp18 | YGR006w | U5 snRNP protein; second step factor |
| Prp17, Cdc40 | YDR364c | Second step factor |
| Brr1 | YPR057w | snRNP biogenesis or recycling |
| Upf3 | YGR072w | Nonsense Mediated Decay |
| Dbr1, Prp26 | YKL149c | Debranching Enzyme |
| Hsp104 | YLL026w | Recovery of splicing from heat shock |
Prp4-1a Strains
prp4-1 mutant: SRY4-1a MATa prp4-1 ade2-101 his7 ura3-52
wild type: SS330 MATa ade2-101 his3-d200 tyr1 ura3-52
Growth Conditions
Cells were grown in rich medium (YEPD) at 26º C overnight then resuspended in 100ml fresh media and allowed to grow for 2-3 doublings.
Prp4-1 Temperature Shift
Prp4-1 and wt strains were grown at 26ºC to A600 of ~1.0, then an equal volume of 48ºC media was added to bring the temperature to 37ºC. Both strains were allowed to grow at 37ºC and samples were taken at 0 (before shift), 5, 15, 30, 60, and 120 minutes after shift to restrictive temperature.
Sample Collection and RNA Isolation
Strains were collected at an A600 of 0.5 to 0.6, centrifuged at room temp. for 2 mins, and the pellets were flash frozen in liquid nitrogen and stored at -80ºC. Total RNA was isolated by a hot phenol method (1).
Microarray Construction
Oligonucleotide Probe Selection
Pilot experiments using oligonucleotides from 34-50 nucleotides (nt) long and symmetrically spanning splice junctions, exon-intron junctions, introns and exons indicated that 40 nt provided a reasonable compromise between strength of hybridization signal and specificity under the hybridization conditions of ~0.78 M Na+ and 62°C, at 21-24°C below the predicted Tms (2). Thus the splice junction oligonucleotides were designed to be 40 nt, with the splice junction positioned between residues 20 and 21. This set of 40-mers has predicted Tms distributed around 84.6±3.5 °C, at 1 M Na+.
We chose the remaining intron and exon sequences using custom designed software program called "OligoPicker." The program chooses sequences based on four main criteria: (a) a predicted Tm near 84.6 °C, (b) limited internal secondary structure, (c) a modification of a heuristic that discourages low information content (3), and (d) absence of significant homology with more than one place in the genome as determined by BLAST.
The selected set of intron oligonucleotides have predicted Tms of 83.71±1.67 °C, and the set of exon oligonucleotides have predicted Tms of 84.43±0.83 °C. Including control genes for normalization and elements for monitoring 244 known or suspected introns, we ordered the synthesis of 832 40-mers with 5' amino linker modifications from WebOligos, Inc. (http://www.weboligos.com). Oligonucleotides (70-mers with 5'amino linkers) for all yeast genes were purchased as a set from Operon, Inc. These are designed to have Tms of 74±3°C in 0.1M Na+, which translates to 86.5±3°C in 1M Na+ (2), comparable to the estimated Tms of the set of 40-mers we designed.
Complete sequences for all custom oligos are available by contacting Manny Ares (ares@biology.ucsc.edu).
Microarray Construction
Oligos synthesized with 5' amines were printed onto glass slides at a concentration of 10pmol/ul in 150mM Sodium Phosphate (pH 8.5) using a robot built to specifications from J. DeRisi. Details of the microarrayer are available at http://cmgm.stanford.edu/pbrown/mguide/index.html. In slide format 1, each 40-mer was printed in quadruplicate, resulting in microarrays that contain more than 3400 elements and four fold oversampling. Slide format 2 contained four copies of the set of 40-mers plus 70-mers for about a thousand intronless genes. Format 3 contains two copies of 40-mers plus the complete Operon set.
Slides were purchased from SurModics, Inc. (now available from Motorola, Inc.) and are coated with a three-dimensional polymer that covalently binds free amines. After printing, slides were incubated in a humid chamber for 36-48 hours. Prior to use, residual reactive groups were blocked with 50mM Ethanolamine in 0.1M Tris (9.0)/0.1% SDS at 50ºC for 30 mins. Slides were then washed with 4xSSC/0.1% SDS at 50ºC for 15 mins., rinsed with water and spun dry.
Probe Preparation, Microarray Hybridization, and Data Acquisition
Fluorescently labeled target sequence sample preparation and hybridization were performed as previously described (4) using reverse transcription of 20 ug of total RNA primed with a mixture of oligo dT and random hexamers (5ug oligo dT, 1ug random). Arrays were scanned and analyzed using a commercially available scanning laser microscope (GenePix 4000) and software package (GenePix Pro 3.0) from Axon Instruments (Foster City, CA).
Normalization & Indexing
Data Normalization
To remove labelling bias, we averaged fluor-reversed pairs of experiments for each mutant (5). Normalization of the data was accomplished by the method of Chen (6) using "Norm" a custom written software application. Norm also screens out artificially high ratios on spots of low signal by adding a prior of two standard deviations of the background to both channels before calculating the ratio. Norm allows users to select control spots to use for normalization and automatically flags spots with low intensity, high background, or high saturation. The signals from the coding regions of seven intronless genes expressed at unchanging levels in 80 yeast microarray experiments from Pat Brown's lab (the stoic genes) were used for normalization. These stoic genes (SLY1, YDR189w, vesicle trafficking between ER and Golgi; SEC4, YFL005w, Golgi to plasma membrane transport; VPS45, YGL095c, Golgi to vacuole transport; PEX4, YGR133w, Ubiquitin conjugating enzyme; TAF145, YGR274c, RNA Pol II general transcription factor; RSC2, YLR357w, chromatin remodeling; YAP1, YML007w, jun-like transcription factor) fall into functional classes other than mRNA processing and display a broad range of expression levels. Experiments with whole genome arrays show that normalization factors derived from these stoic genes are representative of the class of all intronless genes in the genome. Log2ratios from the four replicate array elements for each probe and reverse labeled experiments (typically a total of eight measurements) were averaged for each probe and clustered using software written by M. Eisen (available at http://www.microarrays.org/software.html). Reproducibility and noise estimates were made using six different array batches, four independent wild type by wild type comparisons, and multiple cultures of the same mutant strains compared to wild type. Intron signals for some genes expressed at low levels in wild type cells are undetectable in some cases, but become detectable upon splicing inhibition or in the dbr1 mutant. Intron-containing transcripts from genes expressed at high levels are readily detected in growing wild type cells, as expected if a few percent of transcripts from each gene are yet to be spliced. Clustering in which the wild type by wild type experiments are included with the data presented here show that the hsp104 mutant is indistinguishable from wild type.
Intronless (stoic) Genes used for Normalization
| Name | ORF |
|
| Sly1 | YDR189w | Suppressor of ypt1 -- Vesicle Trafficing between ER and Golgi |
| Sec4 | YFL005w | Golgi to Plasma Membrane Transport |
| Vps45 | YGL095c | Golgi to Vacuole Transport |
| Pex4 | YGR133w | Ubiquitin conjugating enzyme; Peroxisome |
| Taf145 | YGR274c | General RNA Pol II transcription factor |
| Rsc2 | YLR357w | Chromatin Remodeling |
| Yap1 | YML007w | jun-like transcription factor |
Splice Junction and Intron Accumulation Index Calculation
The Splice Junction and Intron Accumulation Indexes provide a measure of splicing which is normalized to levels of transcription and decay on a gene by gene basis. The Splice Junction (SJ) Index is the ratio of the mutant/wild type ratios derived from the normalized signals from the splice junction probe to the exon2 probe: SJ Index = SJmut/SJwt divided by E2mut/E2wt, obtained by subtracting the log2ratio of the exon2 probe from the log2ratio of the splice junction probe. The Intron Accumulation (IA) Index is obtained by subtracting the log2ratio of the exon2 probe from the log2ratio of the intron probe. Because probe performance may not be directly related to absolute transcript amounts in every case, the values of these indexes depend idiosyncratically on the sequences of the probes and are not related in a simple way to splicing efficiency. Given that experimental samples are internally referenced, such indexes are as useful for profiling as the ratios from which they are derived. We also calculated the precursor/mature (PM) index, which is obtained by subtracting the log2ratio of the splice junction probe from the log2ratio of the intron probe. This index mimics the unspliced/spliced ratio used in classical splicing studies (7).
RT-PCR
RT PCR validation of microarray data
RT-PCR and Southern blot analysis was carried out on transcripts from nine different genes (7 intron-containing genes: ARP2, COF1, IMD4, POP8, RUB1, TFC3, TUB3, and 2 of the stoic genes used to normalize the array data, TAF145 and SEC4) using RNA from prp17, prp18 and wild type. Phosphorimage analysis of the amounts of PCR products derived from spliced and unspliced RNAs allowed us to calculate values similar to the SJ and IA Indexes derived from the microarray data by normalizing the yield of PCR products to the control genes. Three primers are used in the PCR reaction to separately amplify spliced and unspliced RNA in the same reaction using a common downstream primer in excess. Sizes of the intron and spliced products were designed to be similar in size to decrease the potential for amplification bias. Severe amplification bias can be discounted because we ensured that under conditions where input cDNA is not saturating, the amplification rates as measured over cycles 14 to 22 of the reaction were about the same for each product. An intron-spanning genomic PCR product was used as the probe in each case, allowing more efficient detection of the less abundant intron-containing PCR product. Blots were scanned on a phosphoimager and quantitated using Image Quant software (Molecular Dynamics).
Comparison of RT-PCR and microarray data
Phosphorimager counts for each PCR product taken at a constant cycle number in the linear range were normalized to the average of the two intronless genes to adjust for initial differences in mRNA in the different samples. These values were treated as intensity measures for intron or splice junction array probes and the ratios between mutant and wild type samples were derived. The ratios for total gene-derived RNA were obtained from the ratios of the sums of the spliced and unspliced counts for each gene, in order to mimic the array signal from the exon 2 probe for indexing. Index values were then calculated using these ratios. The PM Index shown corresponds to the conventional method of determining splicing efficiency whereby the counts in unspliced RNA are divided by counts in spliced RNA in the same lane without normalization, and then averaged over cycle number.
Supplementary Figures
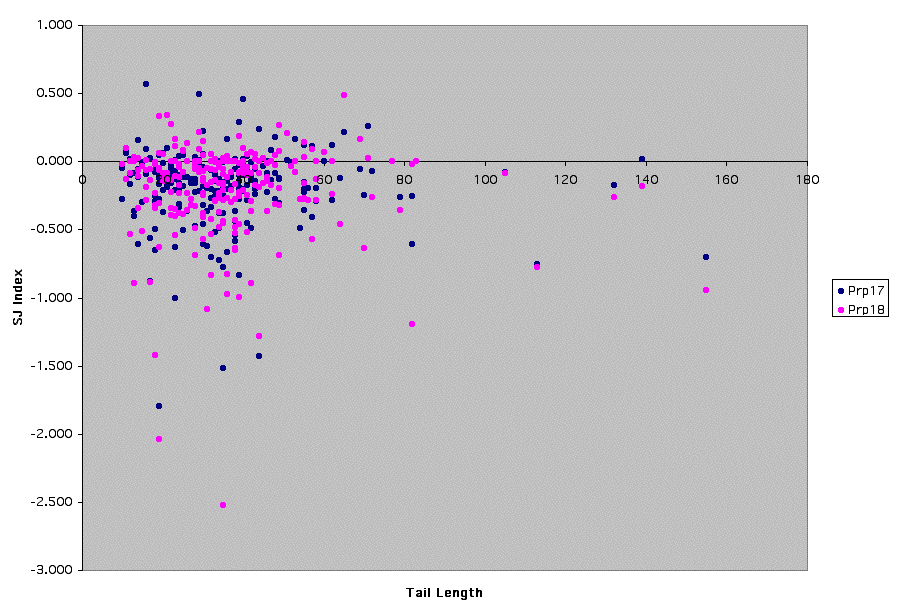
Supplemental Figure 1: Prp17/18 - Graph of SJ Index vs. Distance from Branchpoint to 3'ss
Both Prp17p and Prp18p are involved in the second catalytic step of splicing (8, 9). Using an in vitro splicing reporter construct, Prp18p has been shown to be dispensable for splicing of introns with short branchpoint to 3'ss distances (10). However, the microarray data suggests that Prp17p and Prp18p are not dispensable for removal of introns with short branch point to 3' splice site distances (tail length).
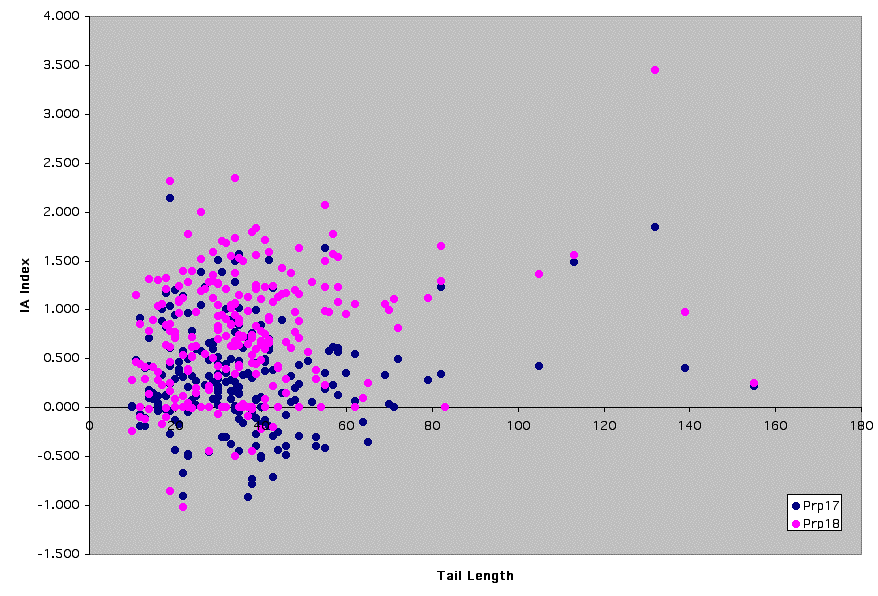
Supplemental Figure 2: Prp17/18 - Graph of IA Index vs. Distance from Branchpoint to 3'ss
See Supplemental Figure 1 for details.
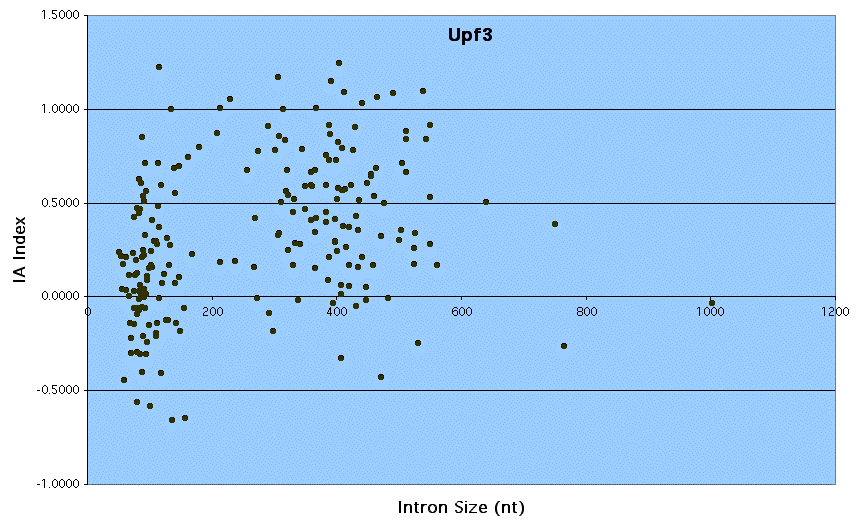
Supplemental Figure 3: Upf3 - Graph of IA Index vs. Intron Size.
Upf3 is involved in nonsense-mediated decay of mRNAs that contain premature stop codons (11). Many introns contain stop codons leading to the expectation that they are subject to nonsense-mediated decay (12, 13). However, levels of unspliced RNA do not accumulate significantly in a upf3 mutant strain (compare upf3 data to dbr1), suggesting that NMD does not contribute greatly to the unstability of pre-mRNA in the cytoplasm.
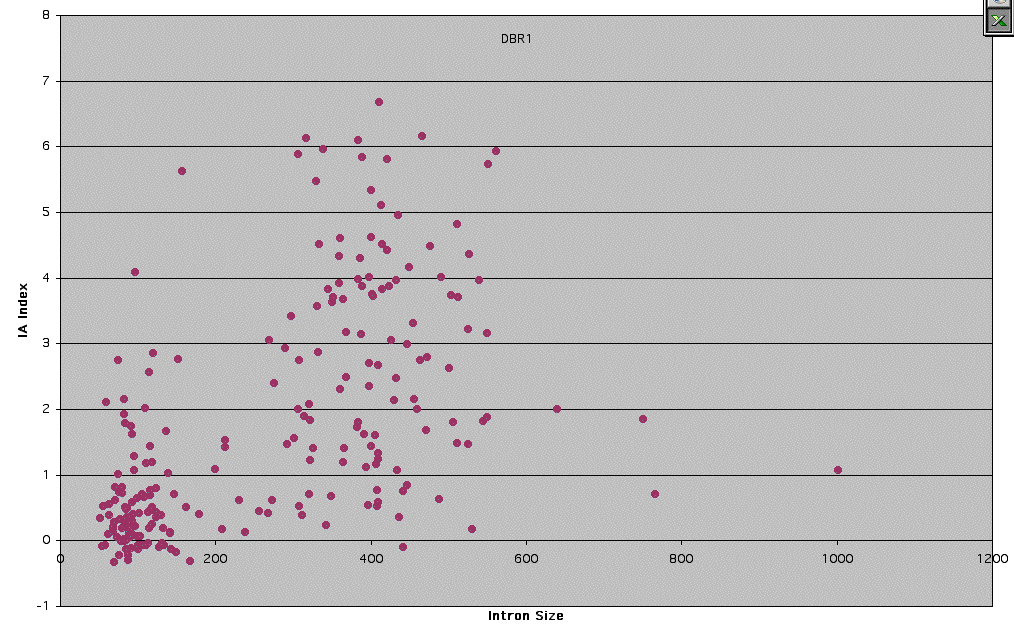
Supplemental Figure 4: Dbr1 - Graph of IA Index vs. Intron Size.
Dbr1p is the debranching enzyme that is responsible for clipping the 2'-5' found in intron lariats which allows for efficient turnover of introns after they have been spliced out (14). Intron accumulation of lariats for snoRNA containing introns showed a correlation with intron size, with smaller introns having increased levels of accumulation (15). Accumulation of intron lariats does not appear to be inversely related to intron size.
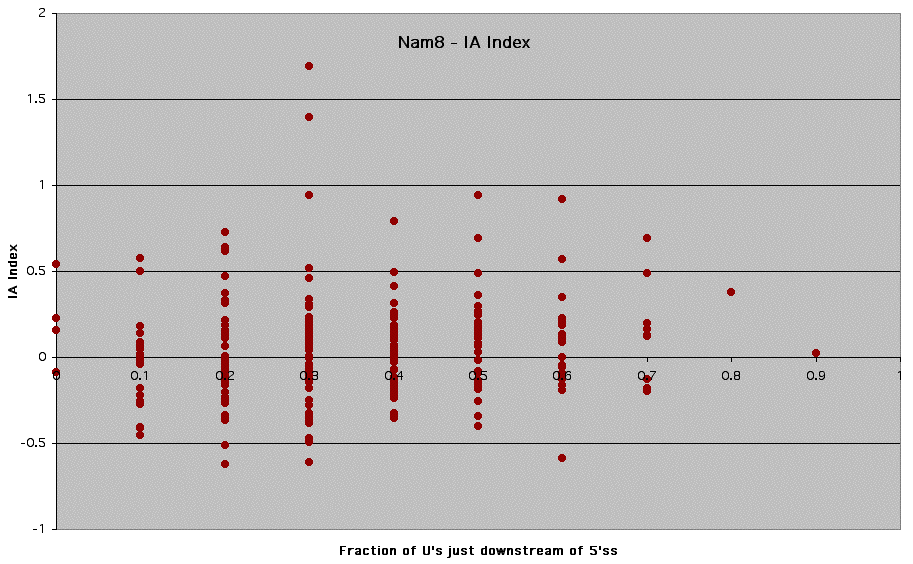
Supplemental Figure 5: Nam8 - Graph of SJ Index vs. Fraction of U's just downstream of 5' ss.
Nam8p is a component of the U1 snRNP that interacts with sequences in the pre-mRNA just downstream of the 5' splice site. Using splicing reporter constructs, Nam8p has also been shown to be required for efficient splicing of introns with non-canonical 5' splice sites (16). Furthermore, the Drosophila homolog of Nam8p, TIA-1 associates selectively with pre-mRNAs that contain U-rich sequences just downstream of the 5' splice site (17).
Supplemental Figure 6: Nam8 - Graph of IA Index vs. Fraction of U's just downstream of 5' ss.
See Supplemental Figure 5 for details.
* Fraction of U's just downstream of 5'ss was calculated using a window of 10 nucleotides immediately following the conserved 5' splice site sequence.
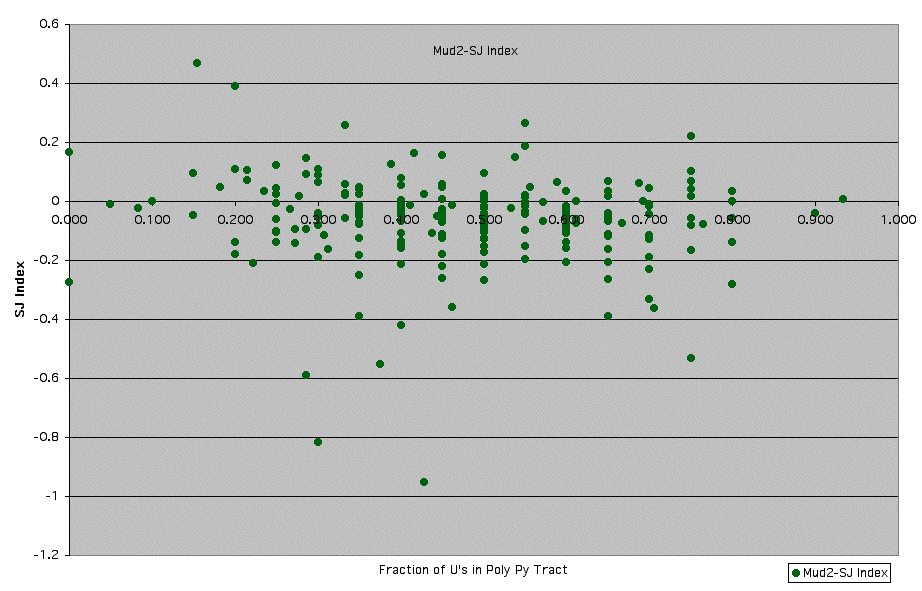
Supplemental Figures 7: Mud2 - Graph of SJ Index vs. Fraction of U's in Poly-Pyrimidine Tract
Mud2p is part of the yeast commitment complex (CC2) and cross-links to pre-mRNA (18). The mammalian homolog of Mud2p, U2AF65 has been shown to interact directly with the polypyrimidine tract located near the 3' end of most mammalian introns (19). Mud2p has also been shown to be involved in the recognition of the nucleotide preceding the conserved UACUAAC branchpoint sequence. Proper recognition of this nucleotide allows for optimal intron recognition (20).
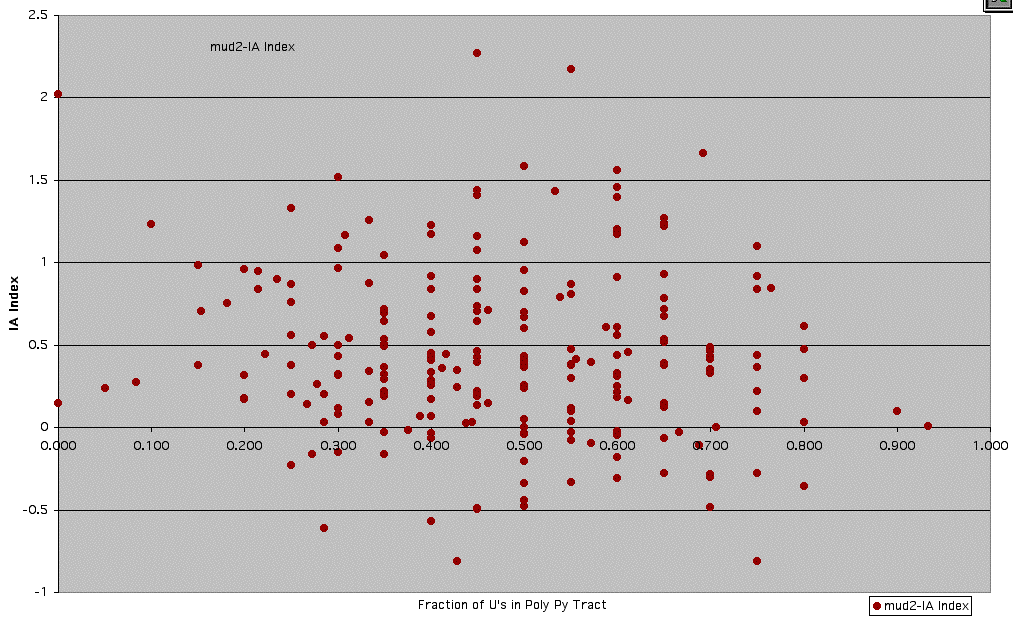
Supplemental Figure 8: Mud2 - Graph of IA Index vs. Fraction of U's in Poly-Pyrimidine Tract
See Supplemental Figure 7 for details.
** Fraction of U's in poly-pyrimide tract was calculated using a window of 20 nucleotides just upstream of the conserved YAG 3' splice site sequence. In cases where there were less than 20 nucleotides between the branchpoint sequence (UACUAAC) and the 3' splice site, a window smaller than 20 nucleotides was used (between the Y of YAG and the last C of UACUAAC).
Supplemental Figure 9: Detection of unspliced RNA in wild type cells
In normal yeast cells, the bulk of RNA from a given gene is the properly spliced, mature mRNA form. Because splicing is typically very efficient, levels of unspliced pre-mRNA are often difficult to detect. The arrays, however, detect unspliced RNA from the vast majority of intron containing genes. One example of this is the MUD1 gene. Mud1p is a non-essential protein that is part of the U1 snRNP. The Mud1 gene also contains an intron. By comparing the Mud1 intron signal in the mud1 deletion strain to the Mud1 intron signal in wild type cells it is clear that we can detect unspliced RNA from the Mud1 gene even under wild type conditions. Mud1 is not a highly transcribed gene. It is estimated by Holstege et al. (21) that Mud1 expression is 0.4 mRNAs per cell.
Microarray Data
The complete data sets are available for download in flat-file tab-delimited text format.
Raw data can be found at the Gene Expression Omnibus (GEO) at http://www.ncbi.nlm.nih.gov/geo/.
GEO Accession Numbers: GSE34, GSE35
All values are average log (base 2) ratios derived from eight
measurements from two reverse labeled experiments.
Derivations of the index calculations can be found in the methods section
under the "Normalization & Indexing" heading.
RNA Processing Factor Knockout Data
Prp4 Temperature Shift Time Series Data
Links
Additional information concerning Yeast Spliceosomal Introns can be found at the Ares Lab Yeast Intron Database.
References
1. Zavanelli MI, Ares M Jr. Genes Dev. 1991 Dec;5(12B):2521-33.
2. SantaLucia J Jr, Allawi HT, Seneviratne PA. Biochemistry. 1996 Mar 19;35(11):3555-62.
3. Lockhart DJ, et al. Nat Biotechnol. 1996 Dec;14(13):1675-80.
4. DeRisi JL, Iyer VR, Brown PO. Science. 1997 Oct 24;278(5338):680-6.
5. DeRisi J, et al. Nat Genet. 1996 Dec;14(4):457-60.
6. Chen Y, et al. J. Biomed. Optics. 1997 2:364-374.
7. Pikielny CW, Rosbash M. Cell. 1985 May;41(1):119-26.
8. Vijayraghavan U, Abelson J. Mol Cell Biol. 1990 Jan;10(1):324-32.
9. Jones MH, Frank DN, and Guthrie C. Proc Natl Acad Sci U S A. 1995 Oct 10;92(21):9687-91.
10. Zhang X and Schwer B. Nucleic Acids Res. 1997 Jun 1;25(11):2146-52.
11. Leeds P et al. Mol Cell Biol. 1992 May;12(5):2165-77.
12. He F et al. Proc Natl Acad Sci U S A. 1993 Aug 1;90(15):7034-8.
13. Lelivelt MJ and Culbertson MR. Mol Cell Biol. 1999 Oct;19(10):6710-9.
14. Chapman KB and Boeke JD. Cell. 1991 May 3;65(3):483-92.
15. Ooi SL et al. RNA. 1998 Sep;4(9):1096-110.
16. Puig O et al. Genes Dev. 1999 Mar 1;13(5):569-80.
17. Forch P et al. Mol Cell. 2000 Nov;6(5):1089-98.
18. Abovich N, Liao XC, and Rosbash M. Genes Dev. 1994 Apr 1;8(7):843-54.
19. Zamore PD and Green MR. Proc Natl Acad Sci U S A. 1989 Dec;86(23):9243-7.
20. Rain JC and Legrain P. EMBO J. 1997 Apr 1;16(7):1759-71.
21. Holstege FC et al. Cell. 1998 Nov 25;95(5):717-28.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}